Compliance & Security

Chinmay Chandgude
Risk Management in Medical Device Development: What Every Startup Should Know

Introducing a medical device to the market is all about safety, reliability, and compliance. Every line of code, every circuit, and every interaction with a patient carries inherent risk. For startups moving fast in MedTech, overlooking structured risk management in medical device development can turn a promising product into a regulatory or financial setback.
According to the FDA, software and design failures account for more than 50% of medical device recalls each year. These recalls don’t just halt operations but erode trust, delay approvals, and raise long-term liability exposure.That’s why global regulators emphasize a formal, traceable approach to identifying and mitigating risks at every stage of medical device development. The ISO 14971:2019 standard, recognized by both the FDA and the European Commission, provides the backbone for how medical device companies should manage risk throughout the product lifecycle.
For early-stage startups, mastering this discipline early doesn’t just prevent costly rework or rejection. It builds a culture of patient safety and product excellence which fundamentally lay the foundation of every successful MedTech brand.
What Is Risk Management in Medical Device Development?
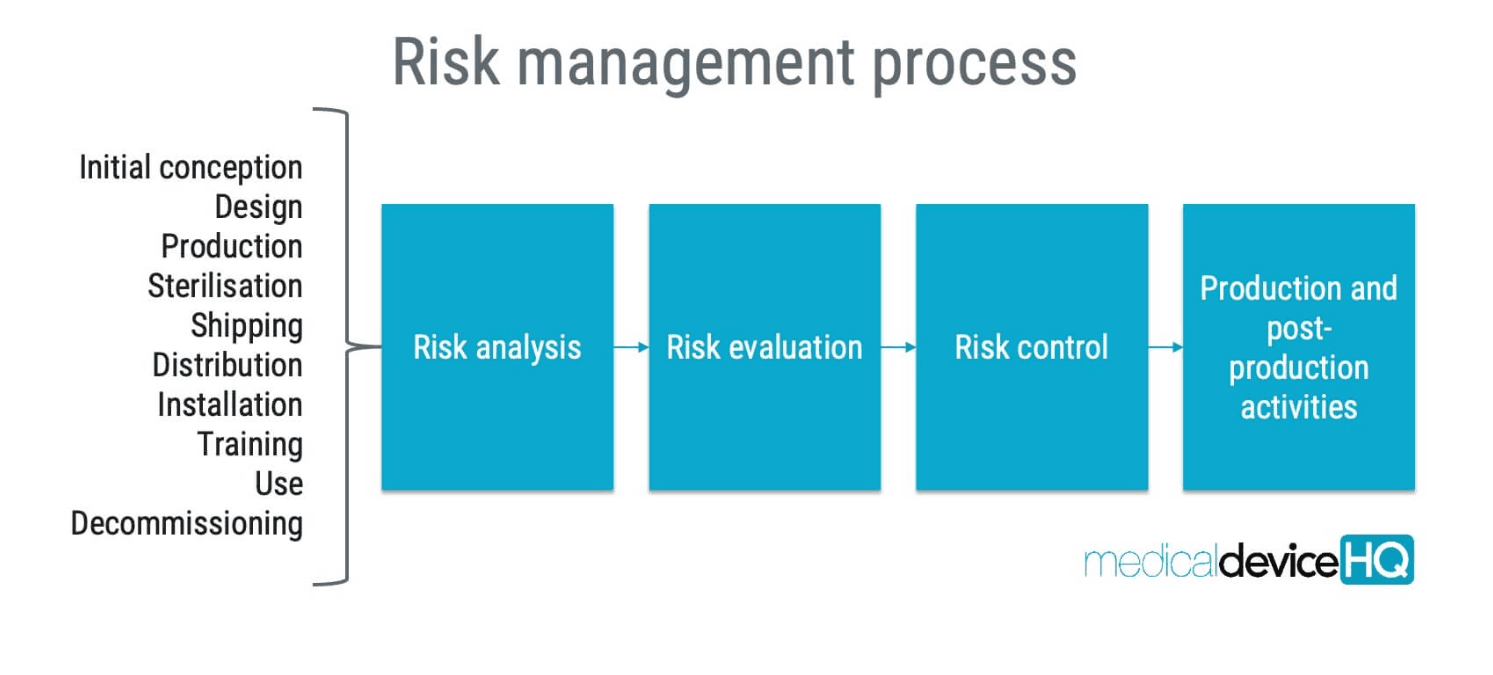
Risk management in medical device development is the ongoing process of identifying, evaluating, controlling, and monitoring anything that could create harm to the patient, the clinician, or the business across the entire lifecycle of the device. This applies to hardware, software, materials, usability, manufacturing, real-world use, and even foreseeable misuse. It starts at concept and continues after launch. ISO 14971 lays out how medical device risk must be managed, and ISO 13485 requires you to build that process into design and development. The FDA’s proposed QMSR is aligned to the same approach. That means you’re not just asked to “think about safety,” you’re required to prove, with documentation, that you understand each hazard, what you’ve done to reduce it, and that those controls actually work.
Done correctly, risk management is not just a compliance checkbox. It’s how you prevent recalls, complaints, and field failures. It forces engineering, clinical, design, and regulatory to align before a weak alarm, confusing UI, or material failure shows up in the hands of a real user. It also creates traceability. Every hazard is linked to a control, every control to a test, every test to evidence. That audit trail is what regulators, partners, and investors expect to see.
For startups, this means embedding traceability and documentation discipline even before the first prototype. By making risk management a design input, not an afterthought, startups can accelerate both regulatory approval and investor confidence.
What Are the Most Common Risk Traps in Medical Device Development?
Even with good intentions, startups often stumble on predictable issues. These five areas account for most delays, recalls, or medical device compliance failures Latent sees in early-stage medical device development projects.
1. Software and Cybersecurity Gaps
More than 50% of medical device recalls are linked to software design or performance errors.
Typical causes include:
Unvalidated algorithms that misinterpret sensor data.
Unpatched vulnerabilities exposing patient information.
Poor version control across test environments
Recommended practice: Implement threat modelling, validation, and penetration testing before release in line with FDA Cybersecurity Guidance (2023).
2. Supply Chain Weaknesses
Relying on single-source suppliers or lacking material traceability often leads to undetected design changes.
Recommended practice: Build a supplier qualification and audit process early, and connect vendor data directly to your design and manufacturing records.
3. Poor Usability and Human Error Risks
Most device misuse happens because interfaces are confusing or instructions are unclear. IEC 62366-1:2015 defines usability engineering principles to reduce these risks.
Recommended practice: Test devices in realistic environments to ensure users understand and operate them safely.
4. Testing and Documentation Gaps
Compressing testing to save time can backfire. Missing test traceability or incomplete documentation leads to audit findings and delayed approvals.
Recommended practice: Treat Verification and Validation as your strongest evidence of safety — not a regulatory hurdle.
5. No Post-Market Monitoring Plan
Neglecting post-market surveillance (PMS) is one of the most common mistakes. Without structured monitoring, early safety signals can go unnoticed, leading to wider recalls and loss of trust.
Recommended practice: Integrate PMS and CAPA (Corrective and Preventive Action) early to maintain medical device compliance and continuous improvement.
How to Integrate Risk Management Early in the Design Phase?
Risk Management for Medical Devices isn’t something you “do just for the audit.” It has to live inside the product, from first requirement to post-market feedback. Teams that treat risk as part of design ship safer devices, clear reviews with less friction, and look credible to regulators and investors.
Here’s how that actually works in practice, based on ISO 14971:2019 and FDA expectations across the full device lifecycle.
1. Start Risk Assessment Early
Risk management begins during concept definition, not validation. At this stage:
Define intended use, user profiles, and operating environments.
Identify potential hazards (mechanical, software, environmental, or usability-related).
Document these in a Risk Management Plan (RMP) aligned with ISO 14971:2019.
2. Build and Maintain the Risk File Continuously
As the design evolves, update the Risk Management File (RMF) continuously. The RMF should connect directly with your:
Design History File (DHF) - how the design evolved.
Verification & Validation (V&V) reports - how risk controls were tested.
Device Master Record (DMR) - how the product will be manufactured.
Every design decision should trace back to a specific hazard and control. This traceability is what regulators look for in FDA 21 CFR 820.30(g) and EU MDR Annex I reviews.
3. Use Structured Tools to Analyze and Prioritize Risks
Risk management only works when it’s systematic. ISO 14971 allows several proven techniques to quantify and prioritize risks.
Use the right tool for the right phase:
Preliminary Hazard Analysis (PHA) – early brainstorming.
Failure Mode and Effects Analysis (FMEA) – evaluate how each component or process might fail.
Fault Tree Analysis (FTA) – trace root causes of system-level hazards.
Hazard Analysis and Critical Control Points (HACCP) – for manufacturing and diagnostic reliability.
These tools help teams focus resources on high-priority risks, reducing costly redesigns later.
4. Integrate Risk Controls into Design and Testing
Every unacceptable risk identified must have a corresponding control measure built into the product. Controls typically include:
Design changes (redundancy, limiters, alarms)
Protective measures (encryption, shielding, labeling)
Training or documentation (clear IFUs and warnings)
Each control must be verified and validated to confirm that it effectively reduces the intended risk without introducing new ones. Verification data belongs in your V&V report and should directly reference the related hazard in your risk file. Digital systems can play a major role here, particularly wearable and IoT-based devices that continuously monitor safety signals.
5. Use Technology to Maintain Traceability
Manual spreadsheets can’t keep up with evolving designs. Modern MedTech teams use digital risk management systems such as:
Greenlight Guru – integrates design and risk controls in one interface.
Jama Connect – links engineering requirements to risk and testing.
MasterControl – automates CAPA and audit documentation.
Using digital tools early prevents version-control errors and ensures your documentation is always audit-ready.
6. Close the Loop - Post-Market Monitoring
Once the device is launched, the work isn’t over. Post-market data including complaints, field failures, cybersecurity incidents, and user feedback. This must feed back into your risk management process.
In practice:
Monitor real-world data through support tickets, connected device telemetry, or clinical feedback.
Update your risk file when new hazards are identified.
Document each Corrective or Preventive Action (CAPA) to show the continuous improvement cycle in action.
This feedback loop: design → verification → field data → redesign - ensures your device remains safe, compliant, and trusted.
7. Make Risk Management a Shared Responsibility
Effective risk management is not the responsibility of one department. It involves:
Engineering - ensuring safety by design.
Regulatory Affairs - ensuring compliance documentation.
Clinical and Usability Teams - ensuring real-world performance.
When risk management is integrated into sprint planning, design reviews, and testing milestones, it becomes a competitive advantage, not a burden.
How Latent Helps You Stay Safe and Compliant
Latent helps MedTech startups build devices and digital health software that are safe, compliant, and built for real-world use:
Compliance-led development from day one: We map potential hazards early, build ISO 14971–aligned risk plans, and maintain traceability across design, verification, and validation.
Safety engineered into the product itself: Our teams integrate software validation, cybersecurity controls, and usability testing into every stage, ensuring devices meet both FDA and EU MDR expectations.
Post-market readiness and surveillance: We implement post-market surveillance and CAPA workflows that feed real-world data directly into design updates, maintaining continuous compliance and long-term product safety.
Introducing a medical device to the market is all about safety, reliability, and compliance. Every line of code, every circuit, and every interaction with a patient carries inherent risk. For startups moving fast in MedTech, overlooking structured risk management in medical device development can turn a promising product into a regulatory or financial setback.
According to the FDA, software and design failures account for more than 50% of medical device recalls each year. These recalls don’t just halt operations but erode trust, delay approvals, and raise long-term liability exposure.That’s why global regulators emphasize a formal, traceable approach to identifying and mitigating risks at every stage of medical device development. The ISO 14971:2019 standard, recognized by both the FDA and the European Commission, provides the backbone for how medical device companies should manage risk throughout the product lifecycle.
For early-stage startups, mastering this discipline early doesn’t just prevent costly rework or rejection. It builds a culture of patient safety and product excellence which fundamentally lay the foundation of every successful MedTech brand.
What Is Risk Management in Medical Device Development?
Risk management in medical device development is the ongoing process of identifying, evaluating, controlling, and monitoring anything that could create harm to the patient, the clinician, or the business across the entire lifecycle of the device. This applies to hardware, software, materials, usability, manufacturing, real-world use, and even foreseeable misuse. It starts at concept and continues after launch. ISO 14971 lays out how medical device risk must be managed, and ISO 13485 requires you to build that process into design and development. The FDA’s proposed QMSR is aligned to the same approach. That means you’re not just asked to “think about safety,” you’re required to prove, with documentation, that you understand each hazard, what you’ve done to reduce it, and that those controls actually work.
Done correctly, risk management is not just a compliance checkbox. It’s how you prevent recalls, complaints, and field failures. It forces engineering, clinical, design, and regulatory to align before a weak alarm, confusing UI, or material failure shows up in the hands of a real user. It also creates traceability. Every hazard is linked to a control, every control to a test, every test to evidence. That audit trail is what regulators, partners, and investors expect to see.
For startups, this means embedding traceability and documentation discipline even before the first prototype. By making risk management a design input, not an afterthought, startups can accelerate both regulatory approval and investor confidence.
What Are the Most Common Risk Traps in Medical Device Development?
Even with good intentions, startups often stumble on predictable issues. These five areas account for most delays, recalls, or medical device compliance failures Latent sees in early-stage medical device development projects.
1. Software and Cybersecurity Gaps
More than 50% of medical device recalls are linked to software design or performance errors.
Typical causes include:
Unvalidated algorithms that misinterpret sensor data.
Unpatched vulnerabilities exposing patient information.
Poor version control across test environments
Recommended practice: Implement threat modelling, validation, and penetration testing before release in line with FDA Cybersecurity Guidance (2023).
2. Supply Chain Weaknesses
Relying on single-source suppliers or lacking material traceability often leads to undetected design changes.
Recommended practice: Build a supplier qualification and audit process early, and connect vendor data directly to your design and manufacturing records.
3. Poor Usability and Human Error Risks
Most device misuse happens because interfaces are confusing or instructions are unclear. IEC 62366-1:2015 defines usability engineering principles to reduce these risks.
Recommended practice: Test devices in realistic environments to ensure users understand and operate them safely.
4. Testing and Documentation Gaps
Compressing testing to save time can backfire. Missing test traceability or incomplete documentation leads to audit findings and delayed approvals.
Recommended practice: Treat Verification and Validation as your strongest evidence of safety — not a regulatory hurdle.
5. No Post-Market Monitoring Plan
Neglecting post-market surveillance (PMS) is one of the most common mistakes. Without structured monitoring, early safety signals can go unnoticed, leading to wider recalls and loss of trust.
Recommended practice: Integrate PMS and CAPA (Corrective and Preventive Action) early to maintain medical device compliance and continuous improvement.
How to Integrate Risk Management Early in the Design Phase?
Risk Management for Medical Devices isn’t something you “do just for the audit.” It has to live inside the product, from first requirement to post-market feedback. Teams that treat risk as part of design ship safer devices, clear reviews with less friction, and look credible to regulators and investors.
Here’s how that actually works in practice, based on ISO 14971:2019 and FDA expectations across the full device lifecycle.
1. Start Risk Assessment Early
Risk management begins during concept definition, not validation. At this stage:
Define intended use, user profiles, and operating environments.
Identify potential hazards (mechanical, software, environmental, or usability-related).
Document these in a Risk Management Plan (RMP) aligned with ISO 14971:2019.
2. Build and Maintain the Risk File Continuously
As the design evolves, update the Risk Management File (RMF) continuously. The RMF should connect directly with your:
Design History File (DHF) - how the design evolved.
Verification & Validation (V&V) reports - how risk controls were tested.
Device Master Record (DMR) - how the product will be manufactured.
Every design decision should trace back to a specific hazard and control. This traceability is what regulators look for in FDA 21 CFR 820.30(g) and EU MDR Annex I reviews.
3. Use Structured Tools to Analyze and Prioritize Risks
Risk management only works when it’s systematic. ISO 14971 allows several proven techniques to quantify and prioritize risks.
Use the right tool for the right phase:
Preliminary Hazard Analysis (PHA) – early brainstorming.
Failure Mode and Effects Analysis (FMEA) – evaluate how each component or process might fail.
Fault Tree Analysis (FTA) – trace root causes of system-level hazards.
Hazard Analysis and Critical Control Points (HACCP) – for manufacturing and diagnostic reliability.
These tools help teams focus resources on high-priority risks, reducing costly redesigns later.
4. Integrate Risk Controls into Design and Testing
Every unacceptable risk identified must have a corresponding control measure built into the product. Controls typically include:
Design changes (redundancy, limiters, alarms)
Protective measures (encryption, shielding, labeling)
Training or documentation (clear IFUs and warnings)
Each control must be verified and validated to confirm that it effectively reduces the intended risk without introducing new ones. Verification data belongs in your V&V report and should directly reference the related hazard in your risk file. Digital systems can play a major role here, particularly wearable and IoT-based devices that continuously monitor safety signals.
5. Use Technology to Maintain Traceability
Manual spreadsheets can’t keep up with evolving designs. Modern MedTech teams use digital risk management systems such as:
Greenlight Guru – integrates design and risk controls in one interface.
Jama Connect – links engineering requirements to risk and testing.
MasterControl – automates CAPA and audit documentation.
Using digital tools early prevents version-control errors and ensures your documentation is always audit-ready.
6. Close the Loop - Post-Market Monitoring
Once the device is launched, the work isn’t over. Post-market data including complaints, field failures, cybersecurity incidents, and user feedback. This must feed back into your risk management process.
In practice:
Monitor real-world data through support tickets, connected device telemetry, or clinical feedback.
Update your risk file when new hazards are identified.
Document each Corrective or Preventive Action (CAPA) to show the continuous improvement cycle in action.
This feedback loop: design → verification → field data → redesign - ensures your device remains safe, compliant, and trusted.
7. Make Risk Management a Shared Responsibility
Effective risk management is not the responsibility of one department. It involves:
Engineering - ensuring safety by design.
Regulatory Affairs - ensuring compliance documentation.
Clinical and Usability Teams - ensuring real-world performance.
When risk management is integrated into sprint planning, design reviews, and testing milestones, it becomes a competitive advantage, not a burden.
How Latent Helps You Stay Safe and Compliant
Latent helps MedTech startups build devices and digital health software that are safe, compliant, and built for real-world use:
Compliance-led development from day one: We map potential hazards early, build ISO 14971–aligned risk plans, and maintain traceability across design, verification, and validation.
Safety engineered into the product itself: Our teams integrate software validation, cybersecurity controls, and usability testing into every stage, ensuring devices meet both FDA and EU MDR expectations.
Post-market readiness and surveillance: We implement post-market surveillance and CAPA workflows that feed real-world data directly into design updates, maintaining continuous compliance and long-term product safety.
Chinmay Chandgude


Chinmay Chandgude is a partner at Latent with over 9 years of experience in building custom digital platforms for healthcare and finance sectors. He focuses on creating scalable and secure web and mobile applications to drive technological transformation. Based in Pune, India, Chinmay is passionate about delivering user-centric solutions that improve efficiency and reduce costs.
Related Posts


Free Interactive Demo
Share your product idea — we'll design your MVP architecture for free, no commitment required. If it's a good fit, we'll show you what building it looks like.